Manufacturing biosimilars
Size matters: biologics are complex
Unlike small-molecule drugs, biologics are large, complex structures that are manufactured in living cells and have significant natural variability.

References:
Adapted from Kozlowski S, Woodcock J, et al. New Engl J Med 2011;365:385–388
Abraham J, Semin Oncol 2013;40:S5–S24
Manufacturing changes occur frequently for biologics
Number of post-approval manufacturing changes to monoclonal antibody therapeutics (accurate as of October 2014)


References:
Adapted from Vezer B, Buzáz S, et al. Curr Med Res Opin. 2016;32:829–834
Reference biologics undergo molecular changes due to process changes
- Manufacturing process changes may result in changes to quality attributes2
- Comparability exercises are required when major changes are made to the manufacturing process2
- EMA and FDA have extensive experience in regulating comparability exercises3
- A changed process is typically approved without additional clinical trials, thereby extrapolating based on an analytical comparability exercise3
Changes detected by CEC in pre- and post-change batches of rituximab1
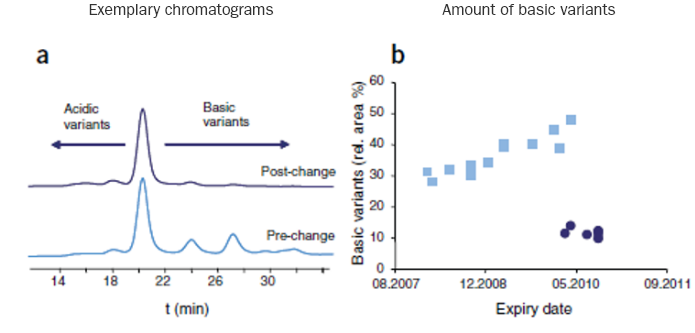
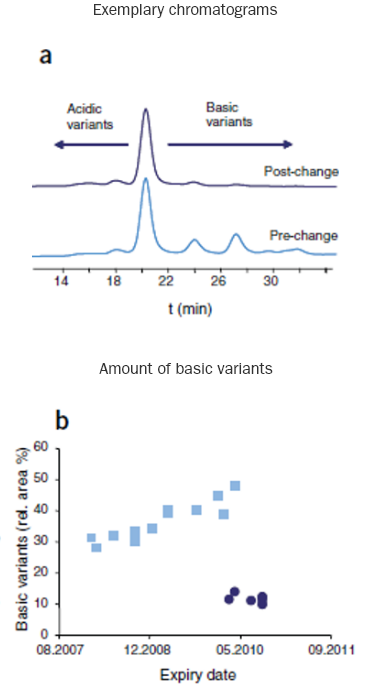
CEC=cation exchange chromatography
References:
Adapted from Schiestl M, Stangler T, et al. Nat Biotechnol 2011;29:310–312;
ICH Guideline Q5E, June 2005. Available from: https://www.ema.europa.eu/en/documents/scientific-guideline/ich-q-5-e-comparability-biotechnological/biological-products-step-5_en.pdf. Accessed 11th November 2019
Declerck P, and Rezk MF. Rheumatology (Oxford) 2017;56:iv4–iv13
Variability is commonplace for all biologics
Number of post-approval manufacturing changes to monoclonal antibody therapeutics (accurate as of October 2014)

Variability is inherent to biologic manufacturing:
no two lots are the same even when the process is “unchanged”1,2
References:
Adapted from Ramanan S and Grampp G. BioDrugs 2014;28:363–372;
Schiestl M, Stangler T, et al. Nat Biotechnol 2011;29:310–312
Advanced technologies have led to improved characterisation of complex biologicals allowing development of high-quality biosimilars
Procedure
The focus of biosimilars development is not to establish benefit:risk but to demonstrate similarity to reference product.
Replicating highly complex biologics has been made possible due to enormous analytical advances

Various methods contribute to a quality attribute fingerprint of the reference drug

References:
Adapted from: Berkowitz S, Engen JR, et al. Nat Rev Drug Discov 2012;11:527–540;
Adapted from: Lee C, Jeong M, et al. Mabs 2017;9:968–977;
Adapted from: Turner A and Schiel JE. Anal Bioanal Chem 2018;410:2079–2093;
Cho IH, Lee N, et al. MAbs 2016;8:1136–1155
During the manufacture of biologics
Reference product batches are continually analysed to define a product’s Quality Target Product Profile (QTPP)
Trend in HMW for batches of trastuzumab over time

The physicochemical and functional properties of a reference product should be characterised comprehensively and monitored periodically to establish the target quality profile used to demonstrate similarity in analytical attributes.1
References:
Adapted from Kim S, Song J, et al. MAbs 2017;9:704–714.
Once a QTPP is confirmed confirmatory similarity studies are performed

References:
Adapted from Vanderkerckhove K, Seidl A, et al. AAPS J 2018;20:68
Real-world evidence is necessary to address any outstanding questions that remain post‑authorisation
Future optimisation of biologics treatment
Transfer of HCPs’ confidence could improve patient adherence
Results from a non-interventional, retrospective multicentre trial in Germany. 13 dermatologists participated in the study and completed physician questionnaires for 246 patients.

Dependency between HCPs’ agreement to given statements and their patients’ therapy adherence and vice versa.
References:
Adapted from Zschocke I, Ortland C, et al. J Eur Acad Dermatol Venereol 2017;31:1014–20